| |
You are here: MIT OpenLabWare » Protein Caging
» Module Overview
Module Overview
Cells are extremely dynamic and mobile. Their ability to move and communicate is necessary for embryonic development and wound repair, but it can also cause harm, as in the proliferation of cancer. These complicated biological processes are all coordinated by a protein called paxillin. Paxillin works by localizing to focal adhesions, which are anchoring points between the cell and the extracellular membrane. Once it is attached to the focal adhesion, paxillin serves as a docking station for numerous proteins that signal different events upon binding.
The creation of binding sites is controlled through a process called phosphorylation, which adds a phosphate group to an amino acid residue. Phosphorylation serves a powerful regulatory role; simply adding a phosphate group can change the conformation of an enzyme to turn it “on” or “off.” Many proteins have multiple phosphorylation sites (amino acids that can accept a phosphate group), so studying phosphorylation can offer many important insights into complex signaling networks. For example, the p53 gene, which encodes for tumor protein p53, is responsible for regulating cell division. Given its role as the “guardian of the cell,” it is understandable that the protein has 18 different phosphorylation sites. When the contents of a cell are damaged beyond repair, the p53 protein is turned “on” through phosphorylation to signal the cell to undergo apoptosis, or essentially “commit suicide.” Programmed cell death prevents defective cells from proliferating uncontrollably and forming a tumor. Thus, the controlled phosphorylation of p53 plays an important role in preventing cancer.
There are numerous methods for determining the effects of protein
phosphorylation, including gene knockout, RNA interference, and site
directed mutagenesis. These techniques work by preventing an animal or
cell from producing a particular kinase, which is an enzyme that
phosphorylates proteins to turn them “on” or
“off,” and observing the consequences. While these studies can be valuable for understanding the wide-spread effects of a particular kinase, they do not offer much insight into what happens over time after the specific phosphorylation of a single site. In 2003, the Imperiali Group at MIT developed a technique to study the effects of peptide phosphorylation in real time. The method synthesizes a peptide with a phosphorylated serine, threonine, or tyrosine residue, where the phosphate group is “caged,” or inactivated by a 2-nitrophenylethyl group. The peptide is activated, or turned “on,” by removing the protective caging group using a pulse of ultraviolet light. Thus, photoactivation of caged phosphopeptides mimics the action of kinases.
Elizabeth Vogel and Barbara Imperiali sought to expand on the group’s earlier work by synthesizing a full-length caged phosphoprotein. Vogel chose to create three paxillin analogs, which were identical except for the tyrosine residue at position 31 (with a natural tyrosine, phosphotyrosine, or caged phosphotyrosine.) They chose to vary this particular tyrosine residue because the downstream effects of its phosphorylation interested other researchers in the Cell Migration Consortium. Thus, they hoped to provide a tool for these biologists to understand the role of a single phosphorylation event in the big picture of cell migration.
All the paxillin analogs were semisynthesized using a technique called native chemical ligation (NCL), which allows any synthetic peptide to be joined to another peptide or protein fragment through a native peptide bond. The first peptide must have a C-terminal thioester, and the second peptide must have an N-terminal cysteine residue, for the peptide bond to be formed. The chemical reaction is pictured below:
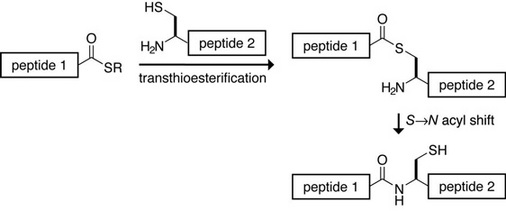
Image courtesy of Prof. Ronald T. Raines.
The semisynthesis of the three paxillin variants involved several steps, which are also documented in the Timeline. Three different N-terminal thioesters were created (each having a different amino acid variant at position 31) and joined to the same C-terminal paxillin fragment using NCL.
- Synthesis of the three 41-residue N-terminal thioesters (representing residues 2-36 of paxillin)
Peptide synthesizers are commonly used to make peptides up to 50 residues in length. Vogel used solid phase peptide synthesis (SPPS) to create three 40 residue peptides, one of which had an
unnatural amino acid of caged phosphotyrosine that they developed in the lab. The carboxylic acid was then converted to a thioester to enable it to be used in the NCL reaction. Finally, the product
was purified using reversed phase high performance liquid chromatography (HPLC) and stored in powder form.
- Synthesis of the C-terminal fragment of paxillin (representing residues 37-557)
- Cloning of the C-terminal fragment of paxillin
This was the first cloning that Vogel had ever done. The DNA encoding for the paxillin fragment and a protease recognition site was transformed into E. coli cells.
- Expression of the C-terminal fragment of paxillin in E. coli
Paxillin is a notoriously challenging protein to express in bacteria, since it is a human protein and has a significant number of rare codons and proline-rich regions. She solved the problem by
using codon-enhanced cells, which are E. coli with an extra plasmid in them that allow them to make more of the rare tRNAs.
- Design of a new construct to increase expression and allow for better purification
To solve problems with poor yields, truncation and degradation, a new construct was designed. It incorporated a GST-fusion protein at the N-terminal, to essentially trick the bacteria into
expressing the protein, and a purification tag at the C-terminal, to remove the truncation and degradation products.
- Cleavage of the fragment from cells
The construct was designed for use with a Factor Xa protease. However, this protease was found to completely degrade the protein through unselective proteolysis. Eventually, TEV, a different
protease, was found to work effectively. A new construct was designed to have a TEV protease cleavage site.
- Testing of Native Chemical Ligation (NCL) technique using a small, simple thioester
In the interest of saving precious quantities of 41 residue N-terminal thioester, a test NCL was performed to join a short thioester with the C-terminal paxillin fragment. The NCL went smoothly
and a proper tag for visualization was determined.
- NCL with actual test thioesters and paxillin fragments
Once the practice ligation was successfully completed, it was time to perform the real ligation using the 41 residue N-terminal thioester. Unfortunately, the ligations consistently failed,
regardless of alterations in the concentrations of the proteins and pH and variations of the thioester. The ligation only worked under very low pH conditions. After emailing Tom Muir, one of the
developers of NCL, it turned out that the glycerol that was being used to store the paxillin fragment in the freezer was capping the cysteine residue and thereby preventing the chemical reaction. The
first glycerol-free attempt at NCL worked perfectly. Next, the procedure was optimized.
- In vitro characterization of semisynthetic paxillin
Several assays were performed to prove the function of the reconstituted paxillin. The assays showed that the semisynthetic paxillin was able to bind to paxillin-binding partners and that it was
recognized by natural kinases.
| |
