| |
You are here: MIT OpenLabWare » Aflatoxin
» Read the Paper
Read the Paper
General Information
This is the HTML version of the paper. Click
here to see a
PDF
file of the paper as it was published in 1977. To view this article at the publisher's website, click here.
Publication Information
Proc. Natl. Acad. Sci. USA
Vol. 74, No. 5, pp. 1870-1874, May 1977
Biochemistry
Structural identification of the major DNA adduct formed by aflatoxin B1
in vitroAbstract The covalent binding of the hepatocarcinogen aflatoxin B1 by rat liver microsomes to calf thymus DNA resulted in a binding level equal to one aflatoxin residue per 60 DNA nucleotides. An aflatoxin derivative-guanine adduct was efficiently liberated from DNA with formic acid. Analytical reversed-phase high-pressure liquid chromatography of the DNA hydrolysate revealed that approximately 90% of the carcinogen bound to DNA could be accounted for as a single component. Preparative high-pressure liquid chromatography was used to isolate sufficient quantities of the adduct for structural analysis from large quantities (340 mg) of DNA. A combination of spectral and chemical data indicates that the major product of the interaction of metabolically activated aflatoxin B1 and DNA is 2,3-dihydro-2-(N7-guanyl)-3-hydroxyaflatoxin B1 with the guanine and hydroxyl functions possessing a trans configuration. The structural data support the hypothesis that the putative 2,3-oxide of aflatoxin B1 is quantitatively important as an intermediate in the binding of aflatoxin B1 to nucleic acids. IntroductionStructural modifications of cellular macromolecules by chemical carcinogens may represent early and requisite events in neoplastic transformation (1, 2). Through interactions of this nature, qualitative changes could be induced in informational macromolecules such as DNA and RNA, and these lesions could provide a molecular basis for alteration of gene expression in carcinogenesis. Identification of the products of these reactions (herein referred to as adducts) is essential in order to: (i) gain insights into mechanisms of carcinogen activation; (ii) determine the reactive centers in these macromolecules; (iii) follow the kinetics of appearance and disappearance of adducts in the cell; and (iv) relate specific patterns of macromolecule modification with the ultimate development of tumors in target organs of susceptible species. Aflatoxin B1 (AFB1) is a very potent liver carcinogen in several animal species (3), and epidemiologic evidence indicates that it is also an important factor in the etiology of human liver cancer in certain sections of the world (4). AFB1 binds covalently to cellular macromolecules, including DNA, in vivo (5-7) and in vitro after metabolic activation (8-10). The relationship of this type of interaction to its mechanism of action has been emphasized (11). Strong indirect evidence has indicated the production of AFB1-2,3-oxide as a major activated metabolite responsible for macromolecular binding in vitro and in vivo (5-7, 9, 12), but structures of specific adducts formed with nucleic acids or proteins have not been determined. The purpose of the research reported here was to determine the structure of the major adduct formed with DNA by AFB1 activated metabolically in vitro. The results indicate that approximately 90% of the binding in vitro can be attributed to a single adduct, which was isolated in sufficient quantity for structural analysis and identified as 2,3-dihydro-2-(N7-guanyl)-3-hydroxyaflatoxin B1 (structure I). 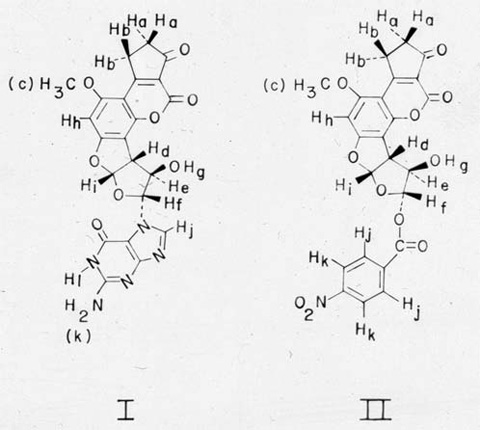 Materials and MethodsLiver microsomes used for metabolic activation of AFB1 were prepared from phenobarbital-treated male Fischer rats (13) by the procedure of Kinoshita et al. (14). The incubation mixture (400 ml) for the binding of AFB1 to DNA included Tris•HC1 (pH 7.5, 45 mM), MgC12 (3 mM), glucose-6-phosphate (5 mM), NADP (0.8 mM, Sigma Chemical Co.), glucose-6-phosphate dehydrogenase (0.4 unit/ml, Sigma Chemical Co.), approximately 1 mg of microsomal protein per ml, calf thymus DNA (20 A260 units/ml or a total of 340 mg; type I, Sigma Chemical Co.), AFB1 [224 µM added in dimethyl sulfoxide; isolated and purified by the method of Asao et al. (15)], and [3H]AFB1 (0.4 mCi, Moravek Biochemicals). The incubation mixture was shaken at 37° for 90 min, and the reaction was terminated by addition of 32 ml of 4 M NaCl/80 ml of 0.17 M sodium dodecyl sulfate and, after 1 min, 512 ml of phenol/chloroform/isoamyl alcohol (50:50:1, wt/vol/vol). After shaking, the emulsion was broken by centrifugation, and DNA was recovered from the aqueous epiphase by ethanol precipitation (0°). The DNA fibers were spooled onto glass rods and were rinsed twice with benzene, once with chloroform, once with ethanol, and twice with diethyl ether. Bound adduct was removed from 340 mg of DNA by incubation in 20 ml of formic acid (88%) at ambient temperature for 60 min. After addition of 80 ml of water, the solution was lyophilized at 0.1 torr (13 Pa). The sample was reconstituted to 100 ml with 10% methanol and held at 0° for 4 hr before centrifugation at 13,000 × g for 15 min. Portions of hydrolysate supernatant were submitted to both analytical and preparative reversed-phase high-pressure liquid chromatography (HPLC), using a Micromeritics model 7000 chromatograph equipped with a Waters Associates model 440 detector (254 and 365 nm). Analytical chromatography of DNA hydrolysates (Fig. 1) was performed on a µBondapak C18 column (Waters Associates) eluted at 50° and 1 ml/min with a water/methanol gradient ranging from 10 to 80% methanol over 40 min. During preparative chromatography, large volumes of DNA hydrolysates (e.g., 30 ml) containing liberated adduct were placed directly onto a 70 × 0.8 cm column packed with C18-Corasil B (Waters Associates) through the pumping head of a positive displacement pump (Milton Roy Co., model 196-31) at 0.8 ml/min. After the sample was loaded, the column was connected to the Micromeritics pumping system and eluted at 3 ml/min with 10% methanol until material absorbing at 254 nm was completely removed. The adduct was eluted from the column by programming the eluant composition to 80% methanol over 40 min. The adduct eluted at 30 min and was crystallized from the column effluent (approximately 60% methanol) at 4°. UV spectra of the adduct (I) were obtained on a Cary model 14 recording spectrophotometer: UV max (0.1 M HCI, pH 1.25) 238, 263, 364 nm (e 17,700, 14,100, 16,100). Because of difficulty in obtaining completely dry material for weighing, there may be some error associated with the absolute values of molar absorptivities. The absorbance maxima remained unaltered in alkaline solution (pH 10). A portion of the adduct was methylated as part of the structural analysis. The adduct (1 mg) was treated with 66 mg of dimethyl sulfate in N,N-dimethylacetamide for 6 hr at 25° by a method similar to that of Jones and Robins for methylation of guanosine (16). The methylated adduct was purified by preparative reversed-phase HPLC. After removal of solvent, the product was treated with 0.1 ml of perchloric acid (72%) at 100° for 60 min. (Adduct not subjected to methylation was hydrolyzed under the same conditions to liberate guanine.) The solution was neutralized with 0.47 ml of 3 M aqueous KOH and adjusted to pH 4 with formic acid (88%). After filtration, portions of hydrolysates were submitted to base analysisansform mode and equipped with an internal heteronuclear deuterium lock. The 90 MHz NMR spectrum of compound II was obtained on a Hitachi Perkin-Elmer R-22 spectrometer. 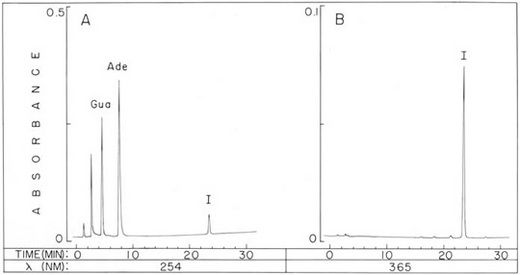
Fig. 1. Analytical HPLC of an aflatoxin-bound DNA hydrolysate. Chromatogram of a formic-acid hydrolysate representing approximately 18 µg of calf thymus DNA incubated in vitro in the presence of AFB1 and rat liver microsomes. The analytical reversed-phase HPLC column was eluted with a water/methanol gradient as described in Materials and Methods, and the detection system monitored absorbance at 254 nm (A) and 365 nm (B). The components Ade, Gua, and I are adenine, guanine, and the adduct, 2,3-dihydro-2-(N7-guanyl)-3-hydroxyaflatoxin B1, respectively. ResultsModification of DNA with AFB1 and Adduct Isolation. The binding in vitro of AFB1 to DNA by rat live microsomes resulted in a level of modification equal to one AFB1 moiety per 60 DNA nucleotides, a level comparable to those obtained with this carcinogen in other systems (8, 10). When microsomal preparations were denatured by heating at 100° for 3 min, no significant binding of AFB1 to DNA occurred. Treatment of unadducted DNA with formic acid at 25° was found to release 66% of the constituent purine bases. The adduct (I), however, was almost completely liberated from adduct-bound DNA by identical treatment, presumably due to destabilization of the glycosidic bond as is characteristic of 7-substituted guanine deoxyribonucleosides (16, 18-20). Analytical HPLC (Fig. 1) revealed that approximately 90% of the radioactivity from the carcinogen originally incorporated into DNA eluted with the adduct peak. Procedures were developed for isolation of sufficient quantities of purified adduct for structure analysis, including large-scale incubations, which provided quantities of adduct in excess of 1 mg. Isolation by analytical HPLC of the adduct from hydrolysates of over 300 mg of DNA was impractical. Consequently, a preparative HPLC system was constructed. In contrast to unmodified nucleic acid components, the adduct is lipophilic and does not migrate significantly when the reversed-phase column is eluted with 10% methanol. Therefore, lyophilized DNA hydrolysates were dissolved in this solvent and large volumes (e.g., 30 ml or the equivalent of over 90 mg of DNA) were delivered onto the column. The adduct accumulated on the column, while more polar components (adenine, guanine, and partially degraded DNA) were eluted. After removal of the adduct from the column with a water/methanol gradient, crystalline material was obtained from the appropriate column effluent fraction by precipitation at 4°. Approximately 5 mg of white crystals were recovered from 340 mg of DNA, and these were pure with respect to other 254 and 365 nm-absorbing components by analytical HPLC. 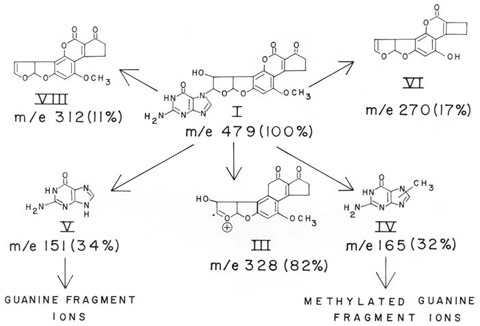
Fig. 2. Proposed structures and fragment ions observed in the field-desorption mass spectrum of adduct (I). The molecular ion at m/e 479 was the base peak; numbers in parentheses are relative abundances of fragment ions. The ions III, IV, V, VI, and VII gave compositions by high-resolution electron-impact mass spectrometry that were attributed to C17H12O7, C6H7N5O, C5H5N5O, C15H10O5, C17H12O6, respectively. Structure Analysis.The structure of the adduct (I) was deduced from a combination of chemical, NMR, UV, and mass spectral evidence. Analysis of the adduct by high-resolution electron-impact (EI) and low-resolution field-desorption (FD) mass spectrometry provided significant structural information (Fig. 2). FD of the adduct resulted in a single molecular ion at m/e 479 which, at higher emitter currents, yielded three additional prominent fragment ions at m/e 328, 165, and 151. Although the molecular ion at m/e 479 was not detected in the EI mode, the latter three ions were measured by high resolution and gave compositions C17H12O7, C6H7N5O, and C5H5N5O, respectively. These can be accounted for as ions III, IV, and V in Fig. 2. Additional fragments observed in the FD spectrum gave compositions determined by EI as C17H12O6 (VII) and C15H10O5 (VI), which are attributable to AFB1 (m/e 312) and the product of O-dealkylation and loss of carbon monoxide from the cyclopentenone ring of AFB1 (m/e 270). The major ion at m/e 328 (III) had the composition of an oxygenated AFB1 produced by the facile cleavage of the adduct bond. The compositions of several other ions measured are similar to those observed (21) in the fragmentation of guanine (V, m/e 151) and a methylated guanine (IV, m/e 165). The partial alkylation of guanine would appear to result from intramolecular transfer following ionization, for no alkylated products were detected at higher mass than the molecular ion at m/e 479. Further evidence for the presence of guanine in the adduct was obtained by chemical
Fig. 3. Determination of the methylated guanine released by hydrolysis of a methylated derivative of the adduct (I). The adduct was methylated with dimethyl sulfate in N,N-dimethylacetamide. The product was purified by preparative reversed-phase HPLC and was hydrolyzed with perchloric acid to cleave the bond between the aflatoxin moiety and the methylated base. The hydrolysate was analyzed by cation-exchange HPLC (A) and the retention time of the major product (11 min) was compared to the retention times of other methylated guanines (B). N7,N9-Dimethylguanine (not shown) eluted at 106 min under these conditions and was not detected in the experiment shown in A. Chromatographic conditions are given in Materials and Methods. 1-MeGua, N1-methylguanine; 2-MeGua, N2-methylguanine; 2-Me2Gua, N2,N2-dimethylguanine; 3-MeGua, N3-methylguanine; 7-MeGua, N7-methylguanine; 9-MeGua, N9-methylguanine. The adduct methylation experiment provided further evidence in support of N7 as the attachment site. The adduct was treated with dimethyl sulfate under conditions that are selective for methylation of the imidazole ring nitrogens (16). A major methylation product (presumed to be N9-methyl-I) was obtained and purified by preparative HPLC. Hydrolysis of the methylated adduct with perchloric acid gave a single methylated guanine, having an HPLC retention time identical to that of authentic N9-methylguanine (Fig. 3A and clearly distinct from other possible methylated guanines that could have been formed under these conditions (Fig. 3B). The identity of N9-methylguanine was further established by isolation (HPLC) and determination of its UV spectrum, which was found to be in-distinguishable from that of authentic N9-methylguanine. Isolation of N9-methylguanine and the absence of detectable methylation at N7 implies an adduct structure in which the guanine base is substituted at N7. A structure with the aflatoxin at any other position (e.g., N3, N2, etc.) would be expected to give significant quantities of N7-methylguanine and, possibly, higher alkylated products under the above methylation and hydrolysis conditions (16, 32). The possibility that N7,N9-dimethylguanine was a significant methylation product was also eliminated by the absence of a peak at the retention time of this base in the methylated adduct hydrolysate chromatogram (not shown in Fig. 3B, because this base eluted at 106 min). Discussion The adduct identified in this study as 2,3-dihydro-2-(N7-guanyl)-3-hydroxyaflatoxin B1 (I) represents the major product formed by the interaction of metabolically activated AFB1 with DNA in vitro. This work extends that of other workers (5-7, 9, 12) in supporting the hypothesis that the proposed AFB1-2,3-oxide is quantitatively important as an intermediate in the binding of the carcinogen to nucleic acids. We have recently found that the in vitro adduct identified in this study is identical chromatographically and in certain spectral characteristics to the major adduct produced in liver DNA when AFB1 is administered to rats (R. G. Croy, unpublished data). This may indicate a functional significance for I, in view of the fact that tissue susceptibility to tumor formation in the rat seems to correlate with the extent of total covalent binding of AFB1 to DNA (7). Alternatively, the situation could be analogous to modification of DNA by alkylating agents, where the pre-dominant modified bases, 7-alkylguanines, are believed to be of lesser importance to tumorigenesis than less abundant products (29, 33). It is noteworthy that the adduct, like the 7-alkylguanines, has a positively charged imidazole ring system in DNA, and it is possible that apurinic sites could be formed spontaneously in the polynucleotide chain as a result of labilization of the glycosidic bond. Additional distortions of the structure of DNA could result from the perturbations by non-polar aflatoxin residues of normal stacking of bases within the double helix. While these factors possibly contribute to a complex alteration of DNA structure, it is difficult to assess their significance as initiating events for carcinogenicity or toxicity, because very little is known about the ways that structural modifications alter the functional properties of nucleic acids. While a sizeable literature exists on the incorporation of alkyl residues (primarily methyl and ethyl groups) into nucleic acids by alkylating agents (20), comparatively few data exist on the interactions of larger functional groups derived from other classes of carcinogens. Weinstein and coworkers have recently identified the products of the reaction of a synthetic 7,12-dimethylbenz[a]anthracene epoxide and a synthetic benzo[a]-pyrene diol epoxide with poly(G), and both compounds were found to bind to the N2 of guanine (26-28). One of the benzo[a]pyrene adduct isomers has identical properties to those of an adduct formed in RNA when this carcinogen is incubated with bovine bronchial explants (27). The carcinogenic arylamine and arylamide, N-methyl-4-aminoazobenzene and 2-acetylaminofluorene, respectively, bind to hepatic nucleic acids predominantly at C-8 of guanine (1, 34, 35), though a minor product recently has been tentatively identified for the latter carcinogen which, like the polycyclic aromatic hydrocarbons, is bound to the N2 of guanine (36). Further studies on identifying the products of interactions of chemical carcinogens with macromolecules should help to clarify the extent to which this phenomenon is related to carcinogenesis. The present research represents the initial results of investigations of the specific products formed when metabolically activated AFB1 reacts with DNA.
Financial support for this research was provided by Grants 5 P01 ES 00597-06 and 5 T32 ES07020-02; and by Contract NO1-CP 43265 from the National Institutes of Health. We express our gratitude to Paul Donahue, Kin Luk, Wayne Siegel, and Mark Goldman for their contributions to this work. We also acknowledge the contributions of Professor Klaus Biemann and the staff of the MIT Mass Spectrometry Facility, which is operated under Grant RR00317 from the Biotechnology Resources Branch, Division of Research Resources of the National Institutes of Health. The 270 MHz NMR spectra were kindly provided by Drs. Leo Neuringer and Warner Harrison of the Francis Bitter National Magnet Laboratory located at MIT and supported by Grant 1-P41-RR00995-01 from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges from funds made available to support the research which is the subject of the article. This article must therefore be hereby marked “advertisement” in accordance with 18 U. S. C. §1734 solely to indicate this fact. - Miller, J. A. (1970) Cancer Res. 30, 559-576.
- Miller, E. C. & Miller, J. A. (1974) in Molecular Biology of Cancer, ed. Busch, H. (Academic Press, New York), pp. 377-402.
- Wogan, G. N. (1973) in Methods in Cancer Research, ed. Busch, H. (Academic Press, New York), Vol. VII, pp. 309-344.
- Wogan, G. N. (1976) in Liver Cell Cancer, eds. Cameron, H. M., Linsell, D. A amp; Warwick, G. P. (Elsevier/North-Holland Biomedical Press, New York), pp. 121-151.
- Swenson, D. H., Miller, E. C. & Miller, J. A. (1974) Biochem. Biophys. Res. Commun. 60, 1036-1043.
- Garner, R. C. & Wright, C. M. (1975) Chem. Biol. Interact. 11, 123-131.
- Swenson, D. H., Lin, J. K., Miller, E. C. & Miller, J. A. (1977) Cancer Res. 37, 172-181.
- Garner, R. C. (1973) Chem. Biol. Interact. 6, 125-129.
- Swenson, D. H., Miller, J. A. & Miller, E. C. (1973) Biochem. Biophys. Res. Commun. 53, 1260-1267.
- Gurtoo, H. L. & Dave, C. V. (1975) Cancer Res. 35, 382-389.
- Miller, J. A. & Miller, E. C. (1976) in Biology of Radiation Carcinogenesis, eds. Yuhas, J. M., Tennant, R. W. & Regan, J. D. (Raven Press, New York), pp. 147-164.
- Swenson, D , Miller, J. A. & Miller, E. C. (1975) Cancer Res. 35, 3811-3823.
- Marshall, W. J. & McLean, A. E. (1969) Biochem. Pharmacol. 18, 153-157.
- Kinoshita, N., Shears, B. & Gelboin, H. (1973) Cancer Res. 33, 1937-1944.
- Asao, T., Büchi, G., Abdel-Kader, M. M., Chang, S. B., Wick, E. L. & Wogan, G. N. (1965) J. Am. Chem. Soc. 87, 882-886.
- Jones, J. W. & Robins, R. K. (1963) J. Am. Chem. Soc. 85, 193-201.
- Beckey, H. D. (1969) Int. J. Mass Spectrom. Ion Phys. 2, 500-503.
- Shapiro, R. (1968) in Progress in Nucleic Acid Research and Molecular Biology, eds. Davidson, J. N. & Cohn, W. E. (Academic Press, New York), Vol. VIII, pp. 73-112.
- Lawley, P. D. & Jarman, M. (1972) Biochem. J. 126, 893-900.
- Singer, B. (1975) in Progress in Nucleic Acid Research and Molecular Biology, ed. Cohn, W. E. (Academic Press, New York), Vol. XV, pp. 219-284.
- Rice, J. M. & Dudek, G. 0. (1967) J. Am. Chem. Soc. 89, 2719-2725.
- Roe, R., Paul, J. S. & Montgomery, P. 0. (1973) J. Heterocycl. Chem. 10, 849-857.
- Büchi, G., Foulkes, D. M., Kurono, M., Mitchell, G. F. & Schneider, R. S. (1967) J. Am. Chem. Soc. 89, 6745-6753.
- Karplus, M. (1963) J. Am. Chem. Soc. 85, 2870-2871.
- Brechbuhler, S., Büchi, G. & Milne, G. (1967) J. Org. Chem. 32, 2641-2642.
- Jeffrey, A. M., Jennette, K. W., Blobstein, S. H., Weinstein, I. B., Beland, F. A., Harvey, R. G., Kasai, H., Miura, I. & Nakanishi, K. (1976) J. Am. Chem. Soc. 98, 5714-5715.
- Weinstein, I. B., Jeffrey, A. M., Jennette, K. W., Blobstein, S. H., Harvey, R. G., Harris, C., Autrup, H., Kasai, H. & Nakanishi, K. (1976) Science 193, 592-595.
- Jeffrey, A. M., Blobstein, S. H., Weinstein, I. B., Beland, F. A., Harvey, R. G., Kasai, H. & Nakanishi, K. (1976) Proc. Natl. Acad. Sci. USA 73, 2311-2315.
- Loveless, A. (1969) Nature 223, 206-207.
- Abola, J. E., Abraham, D. J. & Townsend, L. B. (1976) Tetrahedron Lett. 39, 3483-3486.
- Lawley, P. D. (1966) in Progress in Nucleic Acid Research and Molecular Biology, eds. Davidson, J. N. & Cohn, W. E. (Academic Press, New York), Vol. V, pp. 89-131.
- Jones, J. W. & Robins, R. K. (1962) J. Am. Chem. Soc. 84, 1914-1919.
- Goth, R. & Rajewsky, M. F. (1974) Z. Krebsforsch. 37-64.
- Lin, J. K., Schmall, B., Sharpe, I. D., Miura, I., Miller, J. A. & Miller, E. C. (1975) Cancer Res. 35, 832-843.
- Lin, J. K., Miller, J. A. & Miller, E. C. (1975) Cancer Res. 35, 844-850.
- Westra, J. G., Kriek, E. & Hittenhausen, H. (1976) Chem. Biol. Interact. 15, 149-164.
| |
